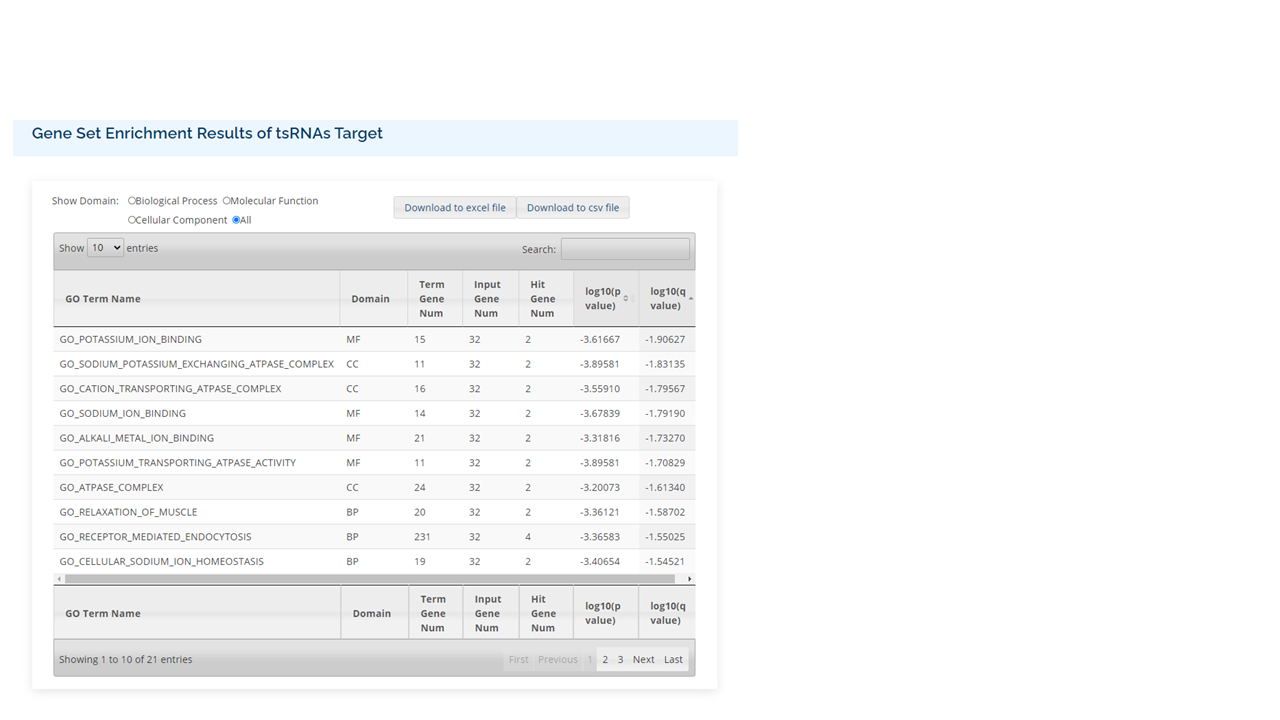
RPM
Different sources of samples could vary in size and depth of sequencing. In order to normalize the samples for direct comparisons, the RPM method is adopted:
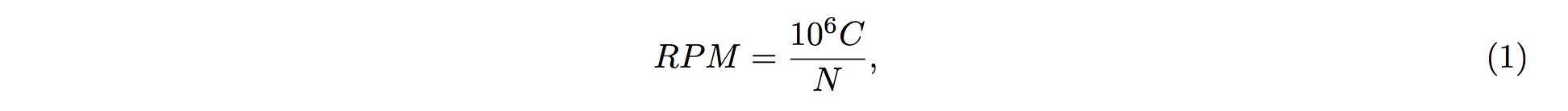
where C is the sum of reads mapped onto one particular position in the tRF, and \( N \) is the sum of reads having been mapped onto the tRNA genes.
p-value
To perform the screening, it is first assumed that fragmented RNAs are randomly distributed along the entire pre-tRNA; that is, the RNAs are uniformly distributed across the entire length of pre-tRNA. According to this assumption, we could conclude that, of the entire length of tRNA, the probability of one small-RNA fragment mapped onto one particular position in the tRNA is
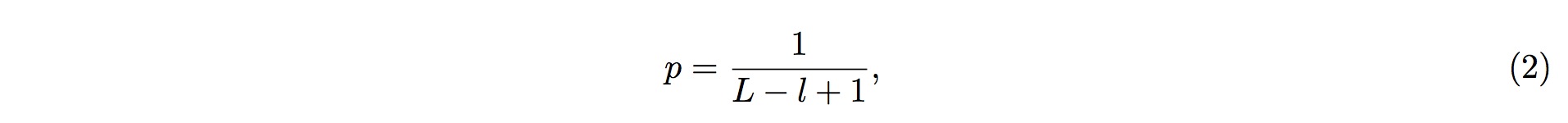
where L is the length (in nucleotide, nt) of the tRNA, and l is the length (in nucleotide, nt) of the small RNA fragment being mapped onto the tRNA.
Therefore, the probability of more than k (inclusive) small RNA fragments mapped onto the same position in the tRNA follows the Binomial distribution, and the probability for this event is
where k is the observed counts of small RNA fragments mapped onto that particular position in the tRNA, and n is the total number of fragments mapped onto the entire tRNA.
If there are more than k (inclusive) small RNA fragments mapped onto one particular position in the tRNA, but the probability of this event occurring by chance (Eq. 2) is less than 1% (referred to the p-value, and can be adjusted to your satisfaction), then we could conclude that the assumption above is false (with 99% confidence, by default); i.e., this event does not occur by chance.
Note, however, that generally tRFs is of more than 16 nt in length. To take this fact into consideration, we should ensure that the tRFs candidate matches with tRNA sequence for consecutive more than 16 nt. (Mismatches, indels are allowed.) This, in turn, corresponds to the requirements that there are at least 16 nt bases in consecutive position in tRNA (which matches with the candidate tRFs sequences) should have a p-values less than 1%. (Figure 1)
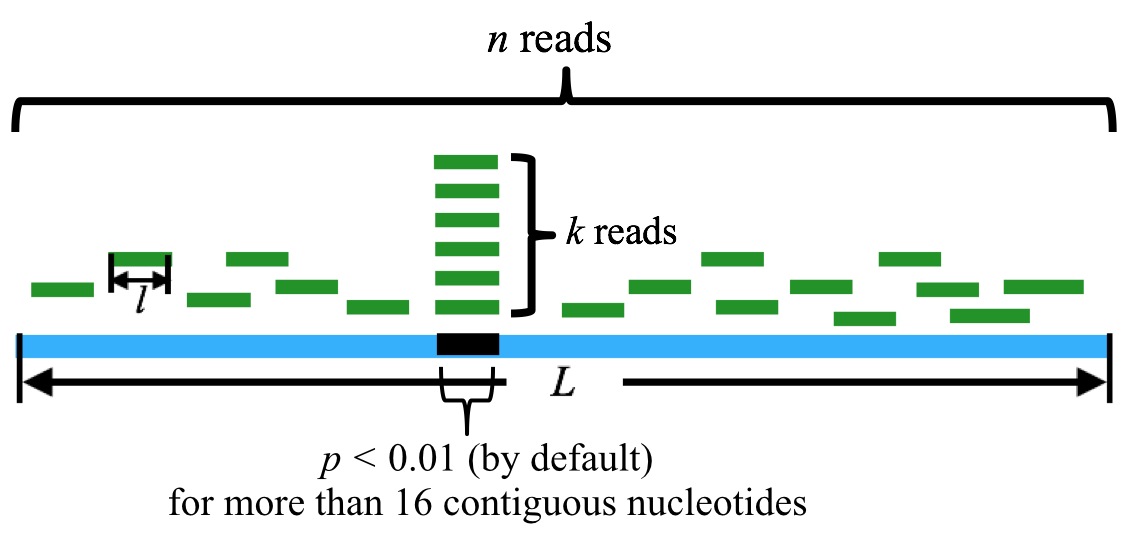
Figure 1. Schematic demonstration of how the core algorithm works.
n is the sum of the reads mapped to the tRNA, k is the sum of the reads that are mapped to the particular position in the tRNA. l is the length of the reads, and L is the length of the tRNA. By default, a tRF candidate corresponds to more than 16 contiguous nucleotides with p-value(s) less than 0.01.
AlignScore
tRFfinder use a scoring scheme to handle different types of mismatches /indels. When the reads are mapped to the tRNA gene sequences and a tRF region is obtained, tRFfinder scores each site of the tRF region according to the following rules (Table 1)
Table 1. The scoring scheme
| Cases on this site of a read |
Score for this site of a read |
| Cases on this site of a read |
Score for this site of a read |
| Perfect match | +1 |
| mismatch or indel of modification sites | -0.5 |
| mismatch or indel on other sites | -1 |
After each site of the tRF region is scored, tRFfinder sums up the scores to get a total score. To eliminate the effect of length on the total score (the longer the region, usually the higher the total score), the total score is divided by the length of the tRF region to get the alignment score for this region. By default, tRFfinder outputs only regions with alignment scores greater than 100 (this threshold can be set by users in the parameter option lists).
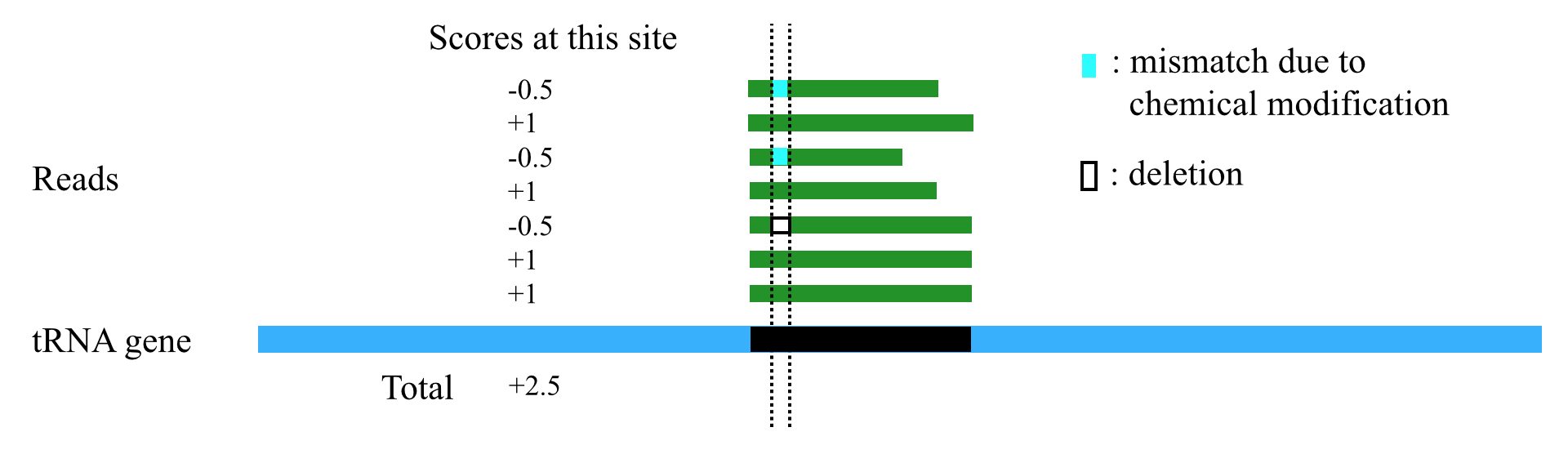
Figure 2. An example of the scoring scheme. The dotted line indicates the potential chemical modification site. When the reads are mapped to the tRNA gene sequences, each site of the tRF region is scored based on the mapped reads. If a mismatch or indel occurs on this site within some reads, a score of -0.5 per read is added to the total score of this site; if a perfect match occurs on this site within other reads, a score of +1 per read is added to the total score.