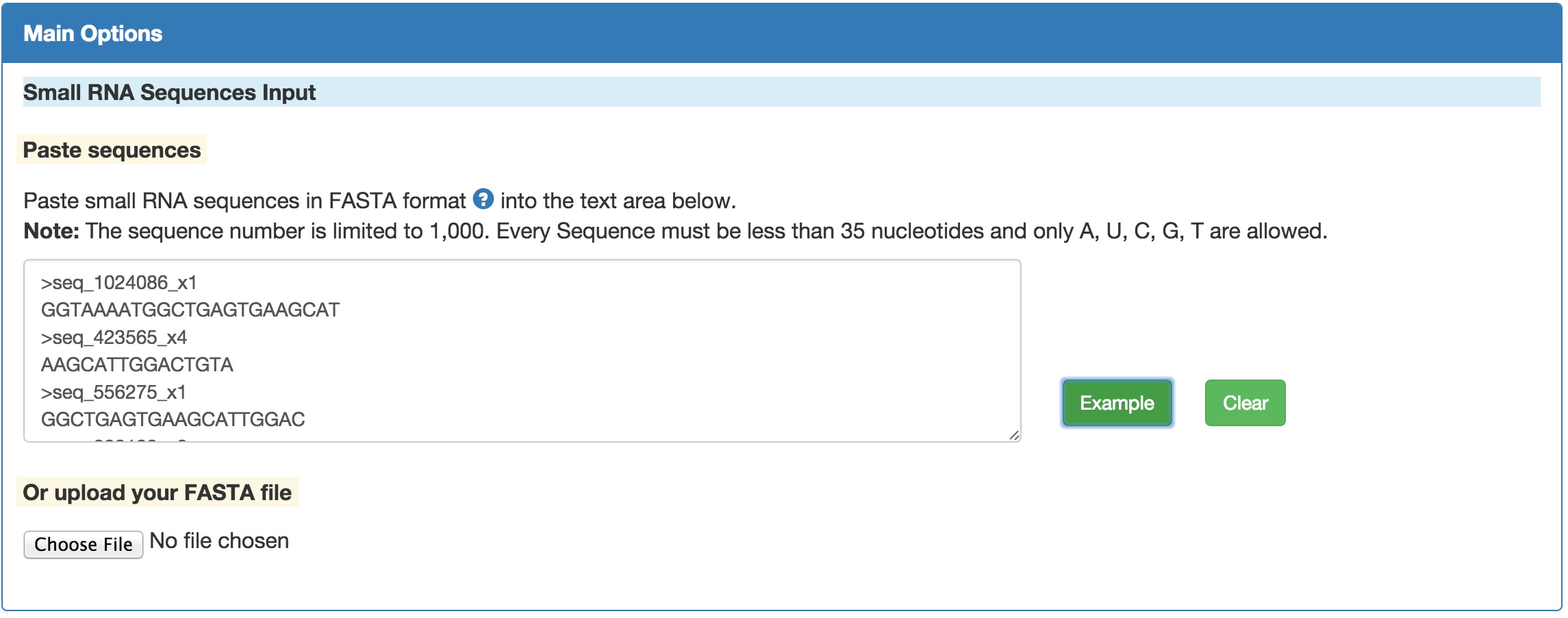
tRFfinder supports only inputs in FASTA format. FASTA is a plain-text format for representing DNA, RNA or protein sequences. Every nucleotide or amino acid is represented by single-letter. For details on FASTA format, please see FASTA Format in Wikipedia.
For tRFfinder, only FASTA fromat for RNA sequences is supported. There are mainly two ways to input your sequence:
-
Paste the sequence(s) into the input box. Be cautious that the sequences must be in FASTA format.
-
Upload the file from your local hard drive. The "Choose File" button
 is for you!
is for you!
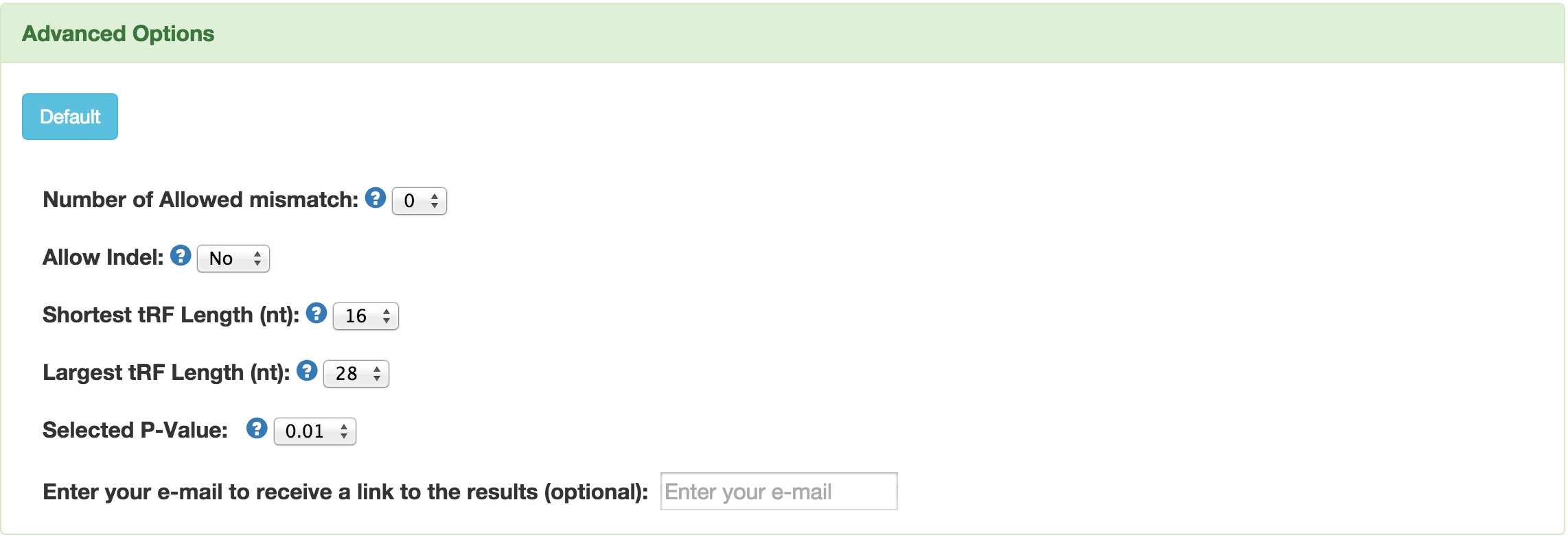
After the input of RNA sequences reads in FASTA format, you can trun to "Advanced Options" for customization of parameters. For an example of input sequenes embedded in the software, please click the  button.
button.
 button at the bottom of the page, you can view the result online. You can also enter your email address in the box above the button, than click
button at the bottom of the page, you can view the result online. You can also enter your email address in the box above the button, than click 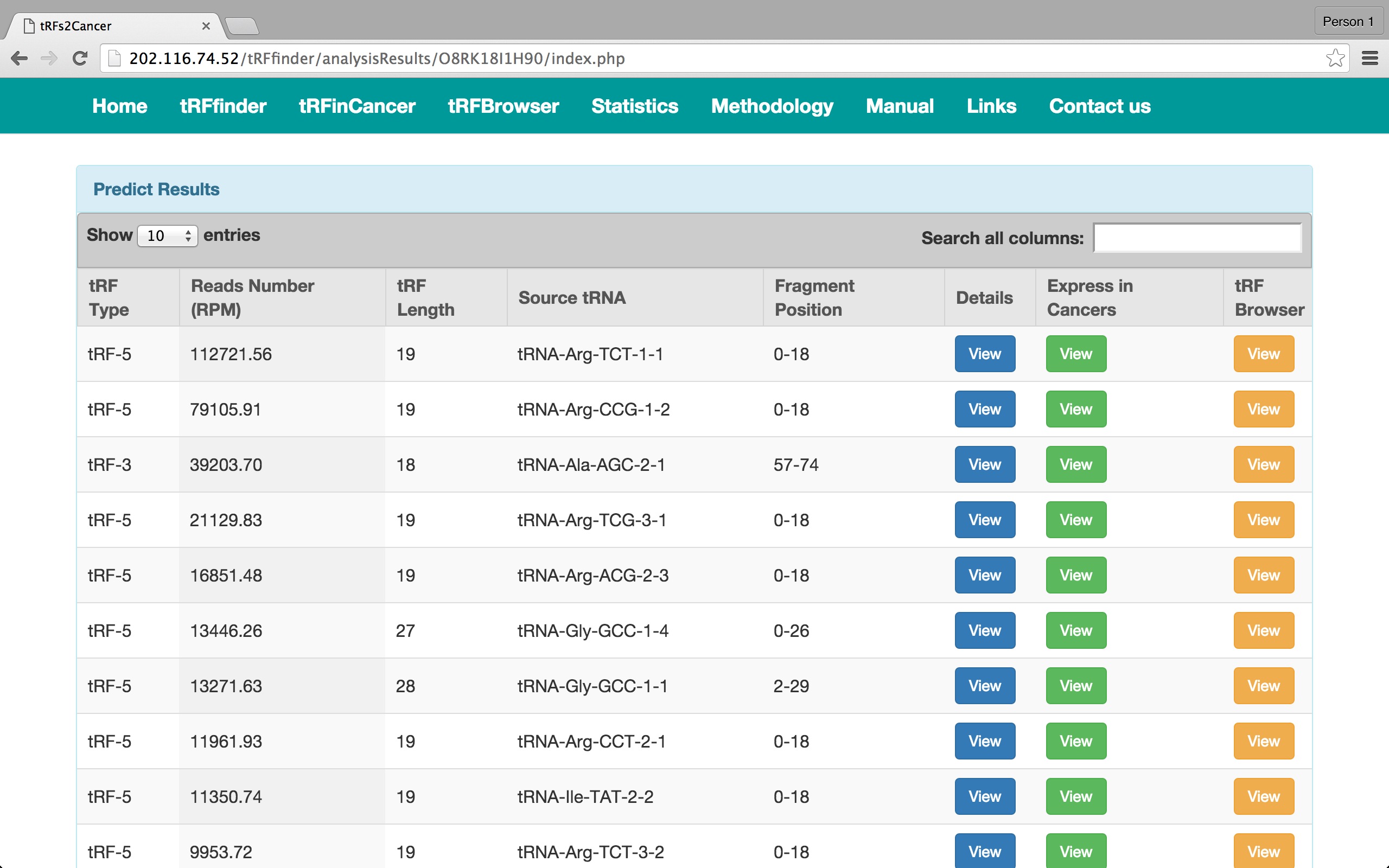
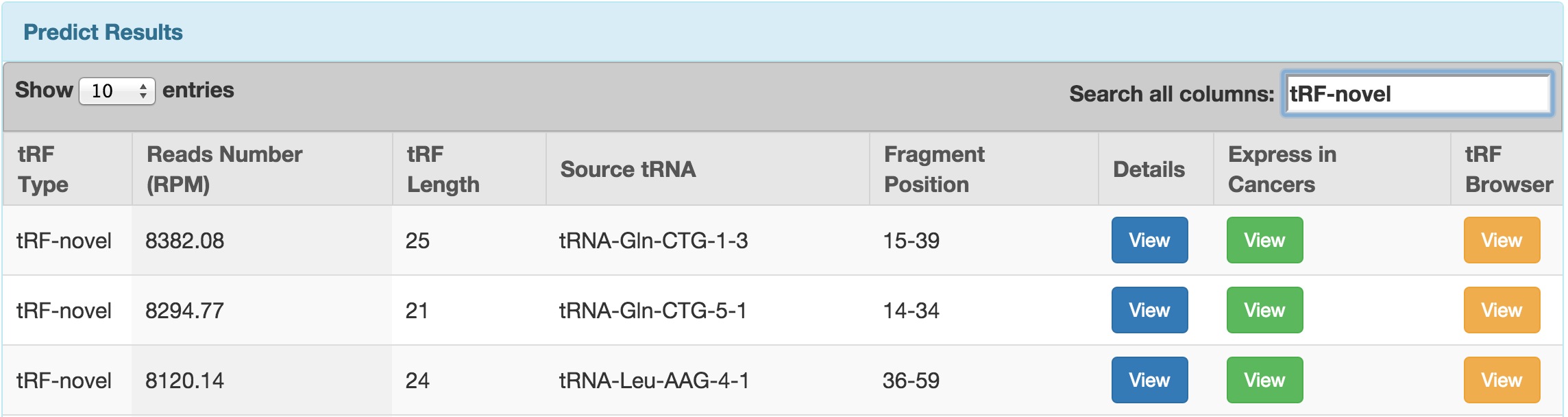
 button. Then come the details of the tRFs. They are divided into three parts.
button. Then come the details of the tRFs. They are divided into three parts.

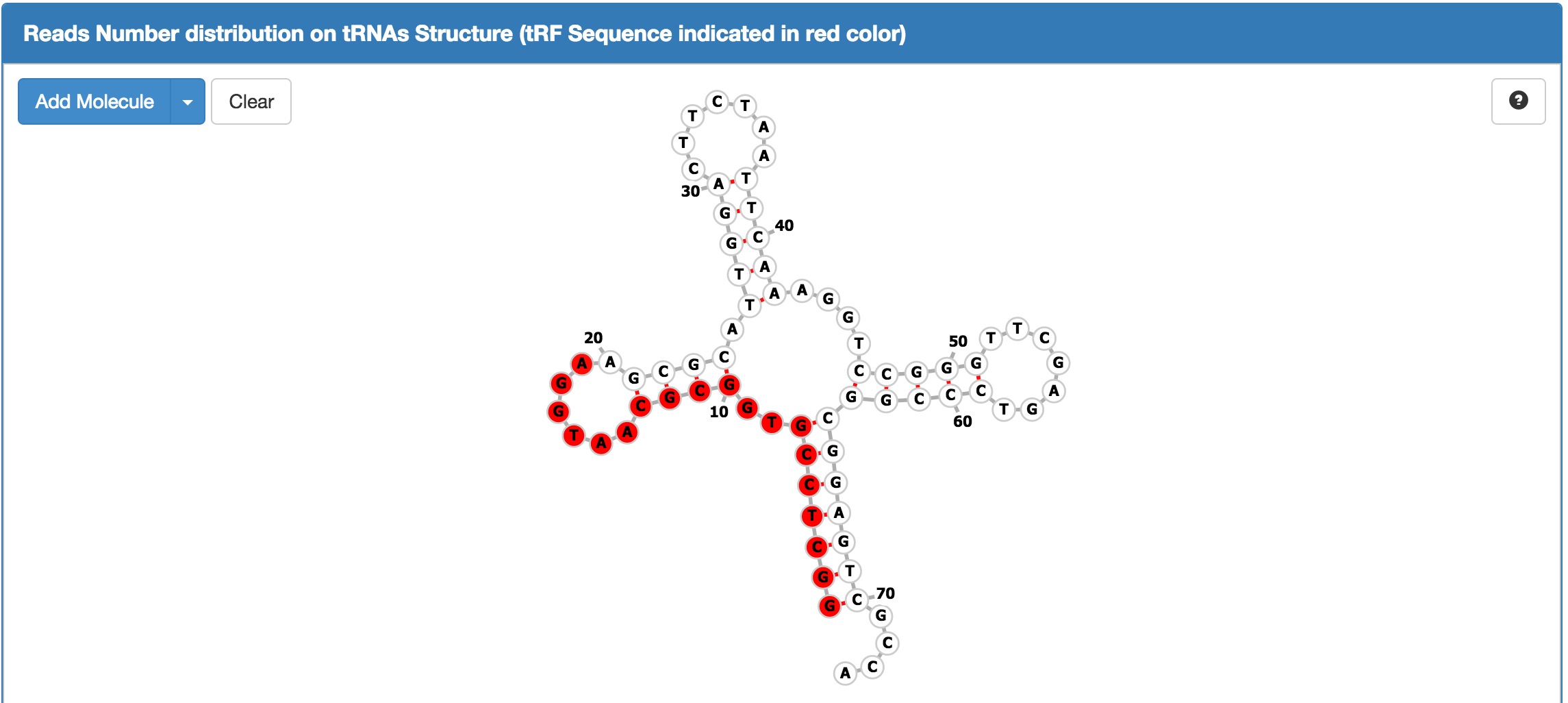
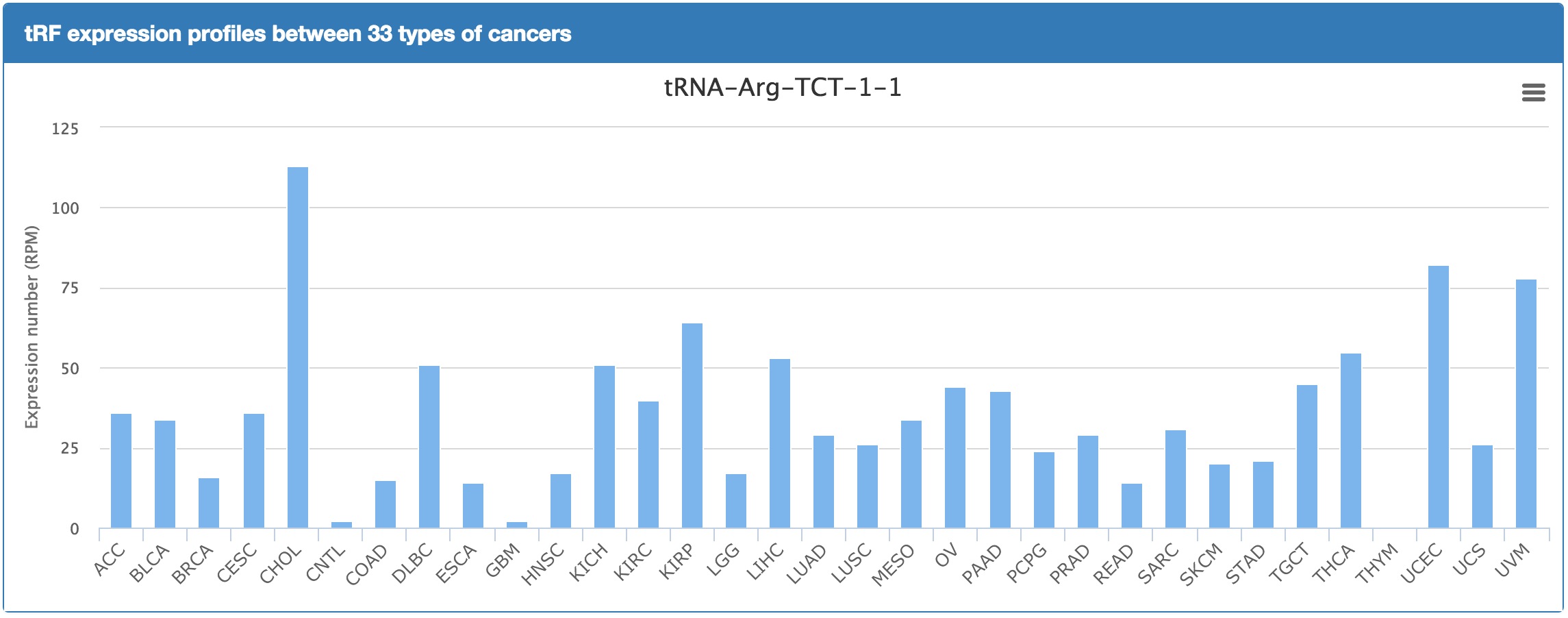
 button in the "Express in Cancers" column. For the manual on this function, see the "tRFinCancer" Section below.
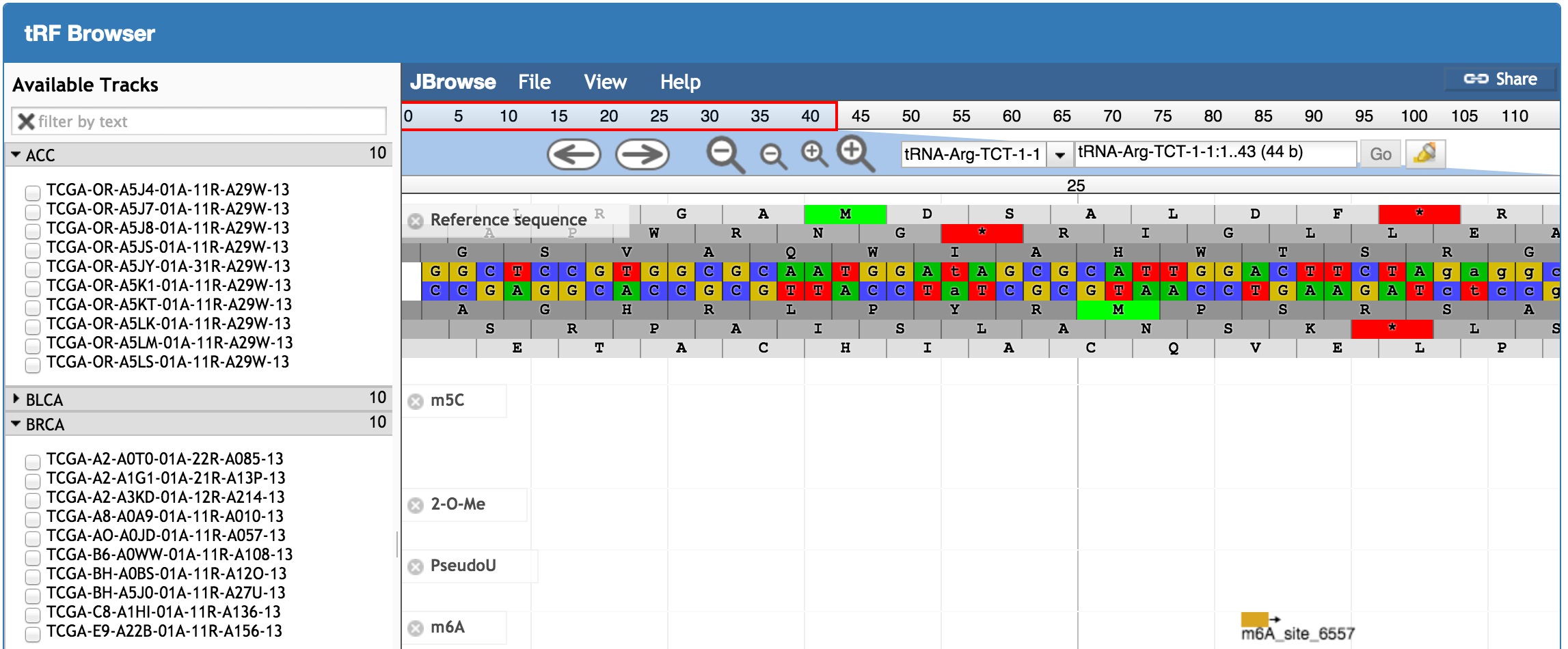
button in the "Express in Cancers" column. For the manual on this function, see the "tRFinCancer" Section below. button. For the manual on this function, see the "tRFBrowser" Section below.
button. For the manual on this function, see the "tRFBrowser" Section below.